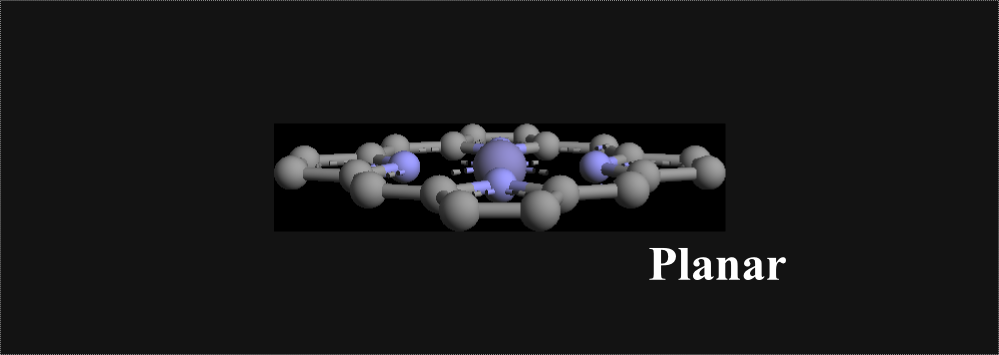
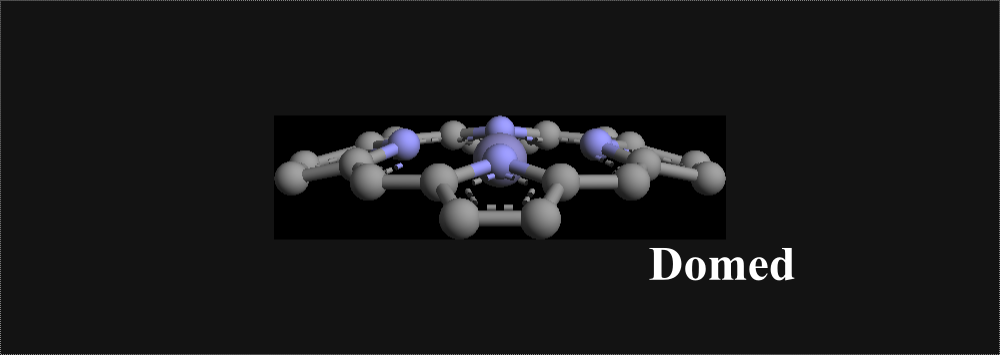
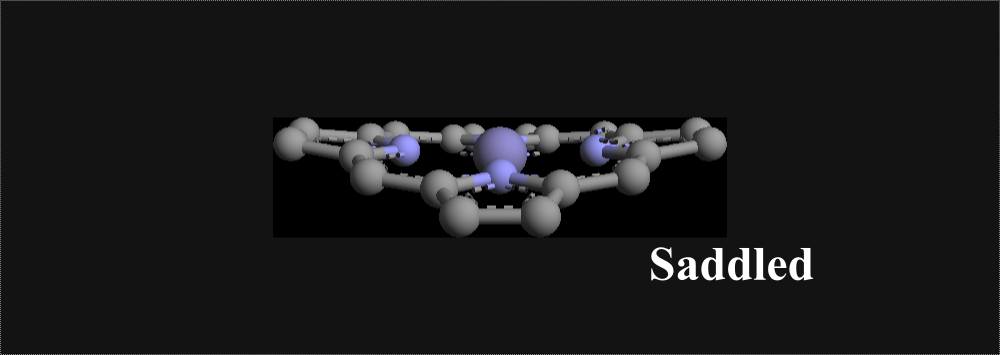
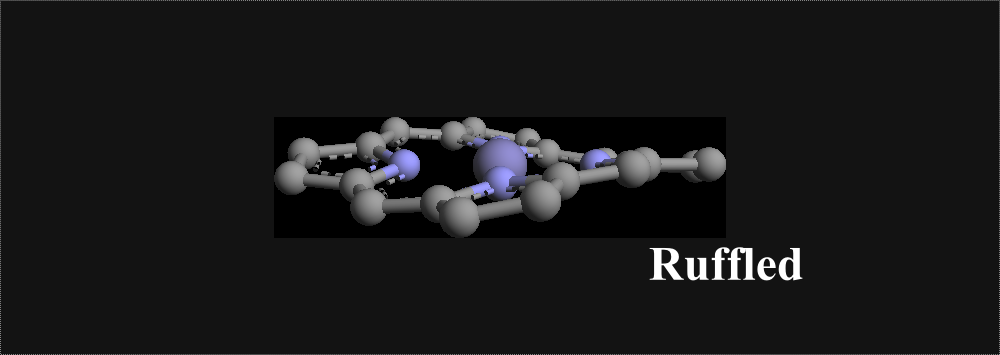
Directions for Using Software
This software is currently able to process two types of files. It processes files downloaded from the Protein Data Bank, and also processes more freely formatted files, with only slight restrictions on how the atomic labels and corresponding cooridinates must appear in the file. We call this second type 'Other data files'. Please see below for how to analyze each type of file.(a) Protein Data Bank (pdb) files.
- Download the pdb file directly from the Protein Data Bank.
- Browse your computer for the file by clicking the button next to where it says 'Choose your input file'.
- Enter the identifier (A, B, C, etc..) of the heme molecule of interest in the field that says 'What is the Chain ID'.
- Click 'Submit'
- Check Table 1 on the next page to make sure that the program correctly read in your input coordinates.
- The output will be given to you in Table 2 on the next page.
Note: If there is more than one heme in the desired chain (e.g. there are two or more hemes bound to the same polypeptide chain), you must delete all heme atoms except for those belonging to the heme of interest from that chain of the pdb file.
(b) Other data files
- Format your file, if necessary, using a text editor of your choice. See below for a description of how data files other than .pdb files must be formatted.
- Browse your computer for the file by clicking the button next to where it says 'Choose your input file'.
- Leave the field 'What is the Chain ID' blank. This is only for use with the Protein Data Bank files.
- Click 'Submit'.
- Check Table 1 on the next page to make sure that the program correctly read in your input coordinates.
- The output will be given to you in Table 2 on the next page.
CHA -2.922045664 -0.430665989 1.503618950
CHB -0.189502240 -2.882752710 -1.681911329
CHC 3.312277465 0.000000000 -0.000000000
CHD -0.201404927 3.320655516 -0.000000000
C1A -2.543161644 -1.415551918 0.597369753
C2A -3.300122384 -2.557503441 0.118039848
C3A -2.536795631 -3.233523649 -0.765698206
C4A -1.273580805 -2.537402754 -0.889087847
C1B 1.092535284 -2.441908525 -1.405189102
C2B 2.378381366 -3.054073579 -1.707678440
C3B 3.339679456 -2.248044119 -1.226447215
C4B 2.717364139 -1.099475528 -0.604882386
C1C 2.716651337 1.249650843 -0.037366932
C2C 3.318932402 2.564868984 -0.156613569
C3C 2.336431982 3.480891908 -0.176156534
C4C 1.073655525 2.782765472 -0.047129018
C1D -1.256487129 2.656283153 0.590309165
C2D -2.391799626 3.253643662 1.244760810
C3D -3.255269257 2.088120765 1.724554070
C4D -2.547876225 0.883957749 1.329175099
NA -1.325409797 -1.432949320 -0.058678088
NB 1.360567479 -1.274479989 -0.736173140
NC 1.356627290 1.435641968 0.030602872
ND -1.384219182 1.272490753 0.671322234
Specifically, you need the atomic label (CHA, NB, C1B, etc...), followed by white space, followed by the x coordinate (in Angstroms) for that corresponding atom, followed by white space, then the y coordinate, then white space, then the z coordinate.
The ordering of the rows does not matter (e.g. CHA can be above or below CHB, etc...).
Your data file can contain extra lines above, below, or between the data lines, as long as those extra lines don't contain the atomic labels (CHA, NB, C1B, etc...).